Structure Modeling
Introduction
Proteins are in the lowest energy conformation. The process in which they assume this conformation from an extended state is called protein folding. It is possible to simulate this event using computers and hence to predict a protein's lowest energy conformation (native state) using protein structure prediction technologies. The reverse, predicting which sequence best fits a specific structure, is also possible, and this is generally referred to as in-silico protein design.
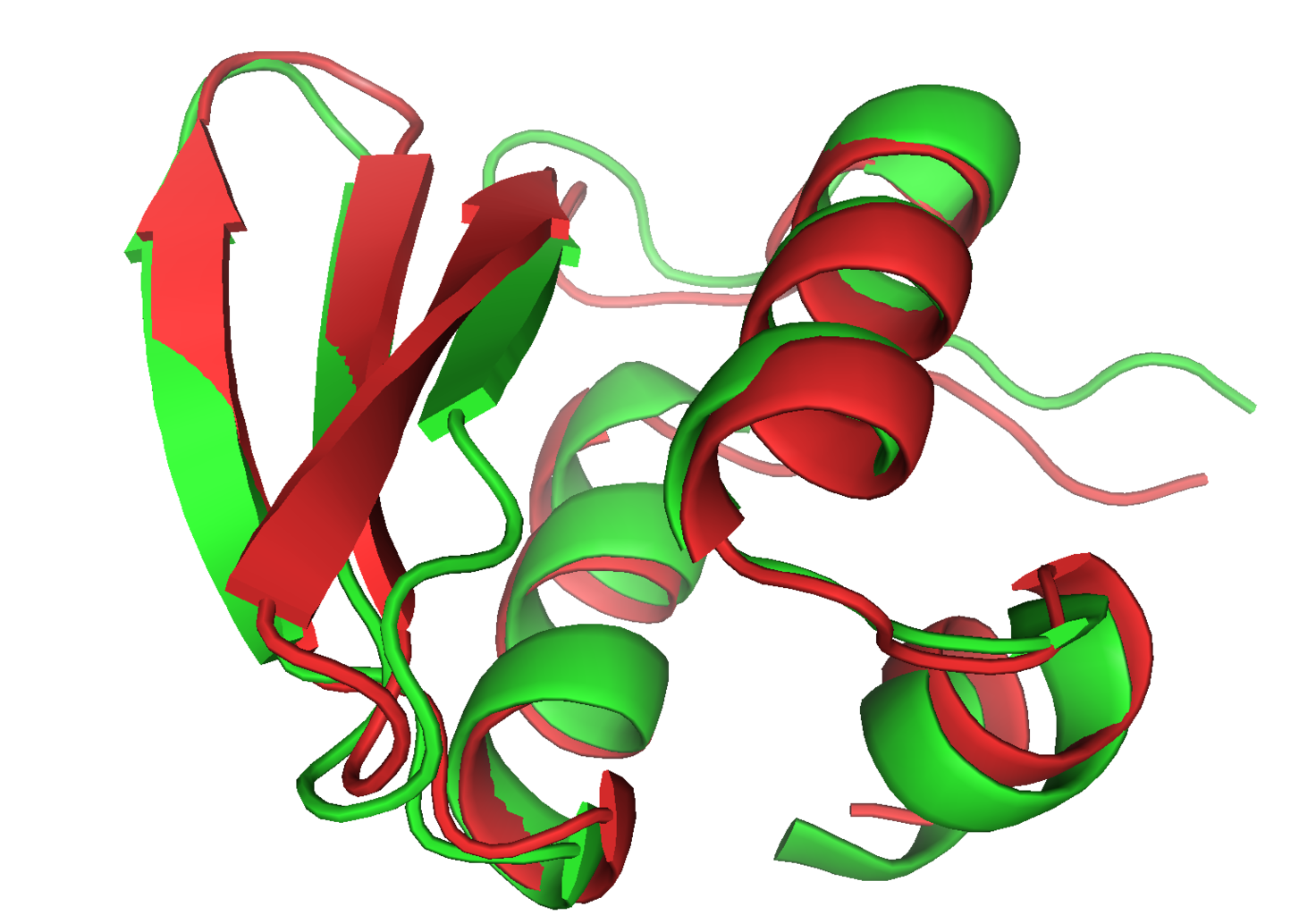
CASP6 model t0281 1 in red is super-imposed on the experimental structure 1whz in green. This near atomic-resolution model was created in a double-blind fashion, and at 1.59A C-alpha RMDS, it was the best prediction for that target.
Research
Rosetta is the software suite we use to predict structures and model protein-protein interactions. I have focused on using protein structure prediction to annotate genomes and to predict molecular functions. It is also possible to characterize proteins with high precision as we did for the DS epimerase. Integrating mass spectrometry (MS) data with de novo protein structure prediction technology can elucidate protein structures fast and cheap with higher accuracy than using only in silico approaches.
References
| Nr. | Reference |
|---|---|
| 17. | Kahraman, Abdullah; Herzog, Franz; Leitner, Alexander; Rosenberger, George; Aebersold, Ruedi; Malmstrom, Lars; Cross-Link Guided Molecular Modeling with ROSETTA. PLoS One (2013), 8: e73411. |
| 16. | Herzog, Franz; Kahraman, Abdullah; Boehringer, Daniel; Mak, Raymond; Bracher, Andreas; Walzthoeni, Thomas; Leitner, Alexander; Beck, Martin; Hartl, Franz-Ulrich; Ban, Nenad; Malmstrom, Lars; Aebersold, Ruedi; Structural probing of a protein phosphatase 2A network by chemical cross-linking and mass spectrometry. Science (2012), 337: 1348-52. |
| 15. | Kunszt, Peter; Malmstrom, Lars; Fantini, Nicola; Subholt, Wibke; Lautenschlager, Marcel; Reifler, Roland; Ruckstuhl, Stefan; Accelerating 3D Protein Modeling Using Cloud Computing. Seventh IEEE International Conference on e-Science Workshops (2011), --: 166-169. |
| 14. | Drew, Kevin; Winters, Patrick; Butterfoss, Glenn; Berstis, Viktors; Uplinger, Keith; Armstrong, Jonathan; Riffle, Michael; Schweighofer, Erik; Bovermann, Bill; Goodlett, David; Davis, Trisha; Shasha, Dennis; Malmstrom, Lars; Bonneau, Richard; The proteome folding project: Proteome-scale prediction of structure and function. Genome Res (2011), 21: 1981-94. |
| 13. | Kahraman, Abdullah; Malmstrom, Lars; Aebersold, Ruedi; Xwalk: Computing and Visualizing Distances in Cross-linking Experiments. Bioinformatics (2011), 27: 2163-4. |
| 12. | Malmstrom, Lars; Goodlett, David; Protein structure modeling. Methods Mol Biol (2010), 673: 63-72. |
| 11. | Malmstrom, Lars; Hou, Liming; Atkins, William; Goodlett, David; On the use of hydrogen/deuterium exchange mass spectrometry data to improve de novo protein structure prediction. Rapid Commun Mass Spectrom (2009), 23: 459-461. |
| 10. | Pacheco, Benny; Maccarana, Marco; Goodlett, David; Malmstrom, Anders; Malmstrom, Lars; Identification of the active site of DS-epimerase 1 and requirement of N-glycosylation for enzyme function. J Biol Chem (2009), 284: 1741-7. |
| 9. | Das, Rhiju; Qian, Bin; Raman, Srivatsan; Vernon, Robert; Thompson, James; Bradley, Philip; Khare, Sagar; Tyka, Michael; Bhat, Divya; Chivian, Dylan; Kim, David; Sheffler, William; Malmstrom, Lars; Wollacott, Andrew; Wang, Chu; Andre, Ingemar; Baker, David; Structure prediction for CASP7 targets using extensive all-atom refinement with Rosetta@home. Proteins (2007), 1: 118-128. |
| 8. | Malmstrom, Lars; Riffle, Michael; Strauss, Charlie; Chivian, Dylan; Davis, Trisha; Bonneau, Richard; Baker, David; Superfamily Assignments for the Yeast Proteome through Integration of Structure Prediction with the Gene Ontology. PLoS Biol (2007), 5: e76. |
| 7. | Kim, David; Chivian, Dylan; Malmstrom, Lars; Baker, David; Automated prediction of domain boundaries in CASP6 targets using Ginzu and RosettaDOM. Proteins (2005), Suppl 7: 193-200. |
| 6. | Chivian, Dylan; Kim, David; Malmstrom, Lars; Schonbrun, Jack; Rohl, Carol; Baker, David; Prediction of CASP6 structures using automated Robetta protocols. Proteins (2005), Suppl 7: 157-66. |
| 5. | Bradley, Philip; Malmstrom, Lars; Qian, Bin; Schonbrun, Jack; Chivian, Dylan; Kim, David; Meiler, Jens; Misura, Kira; Baker, David; Free modeling with Rosetta in CASP6. Proteins (2005), Suppl 7: 128-34. |
| 4. | Cazzanti, Luca; Gupta, Maya; Malmstrom, Lars; Baker, David; Quality Assessment of Low Free-Energy Protein Structure Predictions. Machine Learning for Signal Processing (2005), -: 375-380. |
| 3. | Hazbun, Tony; Malmstrom, Lars; Anderson, Scott; Graczyk, Beth; Fox, Bethany; Riffle, Michael; Sundin, Bryan; Aranda, J; McDonald, W; Chiu, Chun-Hwei; Snydsman, Brian; Bradley, Phillip; Muller, Eric; Fields, Stanley; Baker, David; Yates, John; Davis, Trisha; Assigning function to yeast proteins by integration of technologies. Mol Cell (2003), 12: 1353-65. |
| 2. | Chivian, Dylan; Kim, David; Malmstrom, Lars; Bradley, Philip; Robertson, Timothy; Murphy, Paul; Strauss, Charles; Bonneau, Richard; Rohl, Carol; Baker, David; Automated prediction of CASP-5 structures using the Robetta server. Proteins (2003), 53: 524-33. |
| 1. | Bonneau, Richard; Strauss, Charlie; Rohl, Carol; Chivian, Dylan; Bradley, Phillip; Malmstrom, Lars; Robertson, Tim; Baker, David; De Novo Prediction of Three-dimensional Structures for Major Protein Families. J Mol Biol (2002), 322: 65. |